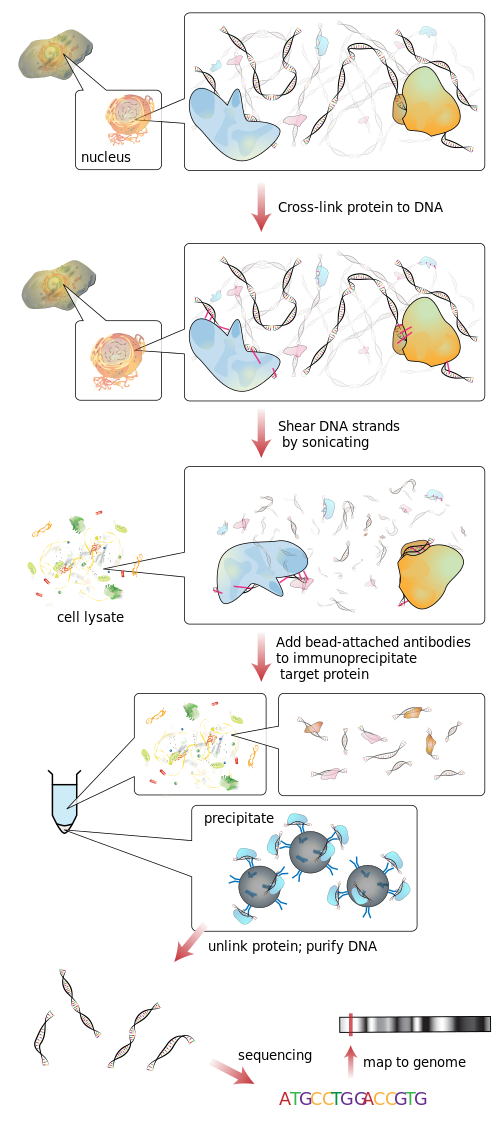
La secuenciación ChIP , también conocida como ChIP-seq , es un método utilizado para analizar las interacciones de proteínas con ADN . ChIP-seq combina la inmunoprecipitación de cromatina (ChIP) con la secuenciación masiva de ADN en paralelo para identificar los sitios de unión de las proteínas asociadas al ADN. Se puede utilizar para mapear con precisión los sitios de unión globales para cualquier proteína de interés. Anteriormente, ChIP-on-chip era la técnica más común utilizada para estudiar estas relaciones proteína-ADN.
Usos
La tecnología ChIP-seq se utiliza principalmente para determinar cómo los factores de transcripción y otras proteínas asociadas a la cromatina influyen en los mecanismos que afectan al fenotipo . Determinar cómo las proteínas interactúan con el ADN para regular la expresión génica es esencial para comprender completamente muchos procesos biológicos y estados patológicos. Esta información epigenética es complementaria al análisis del genotipo y la expresión. La tecnología ChIP-seq se considera actualmente principalmente como una alternativa al ChIP-chip , que requiere una matriz de hibridación . Esto introduce cierto sesgo, ya que una matriz está restringida a un número fijo de sondas. Por el contrario, se cree que la secuenciación tiene menos sesgo, aunque el sesgo de secuenciación de las diferentes tecnologías de secuenciación aún no se comprende por completo. [1]
Los sitios específicos de ADN en interacción física directa con factores de transcripción y otras proteínas se pueden aislar mediante inmunoprecipitación de cromatina . ChIP produce una biblioteca de sitios de ADN objetivo unidos a una proteína de interés. Los análisis de secuencias masivamente paralelas se utilizan junto con bases de datos de secuencias de todo el genoma para analizar el patrón de interacción de cualquier proteína con ADN, [2] o el patrón de cualquier modificación epigenética de la cromatina . Esto se puede aplicar al conjunto de proteínas y modificaciones capaces de ChIP, como factores de transcripción, polimerasas y maquinaria transcripcional , proteínas estructurales , modificaciones de proteínas y modificaciones de ADN. [3] Como alternativa a la dependencia de anticuerpos específicos, se han desarrollado diferentes métodos para encontrar el superconjunto de todas las regiones reguladoras activas de nucleosomas agotados o nucleosomas alterados en el genoma, como DNase-Seq [4] y FAIRE-Seq . [5] [6]
Flujo de trabajo de la secuenciación de ChIP

Chip
ChIP es un método poderoso para enriquecer selectivamente las secuencias de ADN unidas a una proteína particular en células vivas . Sin embargo, el uso generalizado de este método ha sido limitado por la falta de un método lo suficientemente robusto para identificar todas las secuencias de ADN enriquecidas. El protocolo de laboratorio húmedo ChIP contiene ChIP e hibridación. Básicamente, el protocolo ChIP tiene cinco partes [7] que ayudan a comprender mejor el proceso general de ChIP. Para llevar a cabo el ChIP, el primer paso es la reticulación [8] utilizando formaldehído y grandes lotes de ADN para obtener una cantidad útil. Las reticulaciones se realizan entre la proteína y el ADN, pero también entre el ARN y otras proteínas. El segundo paso es el proceso de fragmentación de la cromatina que rompe la cromatina para obtener piezas de ADN de alta calidad para el análisis ChIP al final. Estos fragmentos deben cortarse para que tengan menos de 500 pares de bases [9] cada uno para tener el mejor resultado para el mapeo del genoma. El tercer paso se llama inmunoprecipitación de cromatina, [7] que es la abreviatura de ChIP. El proceso ChIP mejora complejos específicos de proteína-ADN reticulados utilizando un anticuerpo contra la proteína de interés seguido de incubación y centrifugación para obtener la inmunoprecipitación. El paso de inmunoprecipitación también permite la eliminación de sitios de unión no específicos. El cuarto paso es la recuperación y purificación del ADN, [7] que tiene lugar mediante el efecto inverso en el enlace cruzado entre el ADN y la proteína para separarlos y limpiar el ADN con una extracción. El quinto y último paso es el paso de análisis del protocolo ChIP mediante el proceso de qPCR , ChIP-on-chip (matriz híbrida) o secuenciación ChIP. Luego, se agregan adaptadores de oligonucleótidos a los pequeños tramos de ADN que se unieron a la proteína de interés para permitir una secuenciación masiva en paralelo . A través del análisis, las secuencias pueden identificarse e interpretarse por el gen o la región a la que se unió la proteína. [7]
Secuenciación
Después de la selección del tamaño, todos los fragmentos de ADN-ChIP resultantes se secuencian simultáneamente utilizando un secuenciador de genoma. Una sola ejecución de secuenciación puede escanear asociaciones de todo el genoma con alta resolución, lo que significa que las características se pueden ubicar con precisión en los cromosomas. ChIP-chip, por el contrario, requiere grandes conjuntos de matrices de mosaicos para una resolución menor. [10]
Existen muchos métodos de secuenciación nuevos que se utilizan en este paso de secuenciación. Algunas tecnologías que analizan las secuencias pueden utilizar la amplificación de grupos de fragmentos de ADN de ChIP ligados a adaptadores en un sustrato sólido de celda de flujo para crear grupos de aproximadamente 1000 copias clonales cada uno. La matriz de alta densidad resultante de grupos de plantillas en la superficie de la celda de flujo se secuencia mediante un programa de análisis del genoma. Cada grupo de plantillas se somete a una secuenciación por síntesis en paralelo utilizando nuevos nucleótidos terminadores reversibles marcados con fluorescencia. Las plantillas se secuencian base por base durante cada lectura. Luego, el software de recopilación y análisis de datos alinea las secuencias de muestra con una secuencia genómica conocida para identificar los fragmentos de ADN de ChIP. [ cita requerida ]
Control de calidad
ChIP-seq nos ofrece un análisis rápido, sin embargo, se debe realizar un control de calidad para asegurar que los resultados obtenidos son fiables:
- Fracción no redundante : las regiones de baja complejidad deben eliminarse ya que no son informativas y pueden interferir con el mapeo en el genoma de referencia. [11]
- Fragmentos en picos: proporción de lecturas que se encuentran en picos sobre lecturas que se encuentran donde no hay un pico. [11]
Sensibilidad
La sensibilidad de esta tecnología depende de la profundidad de la secuenciación (es decir, el número de etiquetas de secuencia mapeadas), el tamaño del genoma y la distribución del factor objetivo. La profundidad de la secuenciación está directamente relacionada con el costo. Si se deben mapear ligandos abundantes en genomas grandes con alta sensibilidad, los costos son altos ya que se requerirá una cantidad enormemente alta de etiquetas de secuencia. Esto contrasta con ChIP-chip en el que los costos no están correlacionados con la sensibilidad. [12] [13]
A diferencia de los métodos ChIP basados en microarrays , la precisión del ensayo ChIP-seq no está limitada por el espaciamiento de las sondas predeterminadas. Al integrar una gran cantidad de lecturas cortas, se obtiene una localización del sitio de unión altamente precisa. En comparación con ChIP-chip, los datos de ChIP-seq se pueden utilizar para localizar el sitio de unión dentro de unas pocas decenas de pares de bases del sitio de unión de la proteína real. Las densidades de etiquetas en los sitios de unión son un buen indicador de la afinidad de unión proteína-ADN, [14] lo que facilita la cuantificación y comparación de las afinidades de unión de una proteína con diferentes sitios de ADN. [15]
Investigación actual
Asociación de ADN con STAT1: se utilizó ChIP-seq para estudiar los objetivos de STAT1 en células HeLa S3, que son clones de la línea HeLa que se utilizan para el análisis de poblaciones celulares. [16] Luego, se comparó el rendimiento de ChIP-seq con los métodos alternativos de interacción proteína-ADN de ChIP-PCR y ChIP-chip. [17]
Arquitectura de nucleosomas de promotores: utilizando ChIP-seq, se determinó que los genes de levadura parecen tener una región promotora libre de nucleosomas mínima de 150 pb en la que la ARN polimerasa puede iniciar la transcripción. [18]
Conservación de factores de transcripción: se utilizó ChIP-seq para comparar la conservación de los factores de transcripción en el tejido del prosencéfalo y del corazón en ratones embrionarios. Los autores identificaron y validaron la funcionalidad cardíaca de los potenciadores de la transcripción y determinaron que los potenciadores de la transcripción para el corazón están menos conservados que los del prosencéfalo durante la misma etapa de desarrollo. [19]
Secuenciación de ChIP en todo el genoma: se realizó una secuenciación de ChIP en el gusano C. elegans para explorar los sitios de unión de 22 factores de transcripción en todo el genoma. Hasta el 20% de los genes candidatos anotados se asignaron a factores de transcripción. Varios factores de transcripción se asignaron a regiones de ARN no codificantes y pueden estar sujetos a variables ambientales o de desarrollo. También se identificaron las funciones de algunos de los factores de transcripción. Algunos de los factores de transcripción regulan genes que controlan otros factores de transcripción. Estos genes no están regulados por otros factores. La mayoría de los factores de transcripción sirven como objetivos y reguladores de otros factores, lo que demuestra una red de regulación. [20]
Inferencia de la red reguladora: se demostró que la señal ChIP-seq de la modificación de histonas estaba más correlacionada con los motivos de factores de transcripción en los promotores en comparación con el nivel de ARN. [21] Por lo tanto, el autor propuso que el uso de ChIP-seq de modificación de histonas proporcionaría una inferencia más confiable de las redes reguladoras de genes en comparación con otros métodos basados en la expresión.
ChIP-seq ofrece una alternativa a ChIP-chip. Los datos experimentales de ChIP-seq de STAT1 tienen un alto grado de similitud con los resultados obtenidos por ChIP-chip para el mismo tipo de experimento, con más del 64 % de picos en regiones genómicas compartidas. Debido a que los datos son lecturas de secuencias, ChIP-seq ofrece un proceso de análisis rápido siempre que haya una secuencia genómica de alta calidad disponible para el mapeo de lecturas y el genoma no tenga contenido repetitivo que confunda el proceso de mapeo. ChIP-seq también tiene el potencial de detectar mutaciones en secuencias de sitios de unión, lo que puede respaldar directamente cualquier cambio observado en la unión de proteínas y la regulación genética.
Análisis computacional
Al igual que con muchos enfoques de secuenciación de alto rendimiento, ChIP-seq genera conjuntos de datos extremadamente grandes, para los cuales se requieren métodos de análisis computacional apropiados. Para predecir los sitios de unión del ADN a partir de los datos de recuento de lecturas de ChIP-seq, se han desarrollado métodos de llamada de picos . Uno de los métodos más populares [ cita requerida ] es MACS, que modela empíricamente el tamaño del cambio de las etiquetas de ChIP-Seq y lo utiliza para mejorar la resolución espacial de los sitios de unión predichos. [22] MACS está optimizado para picos de mayor resolución, mientras que otro algoritmo popular, SICER, está programado para solicitar picos más amplios, que abarcan kilobases a megabases para buscar dominios de cromatina más amplios. SICER es más útil para las marcas de histonas que abarcan cuerpos de genes. Se puede utilizar un método matemático más riguroso, BCP (Punto de cambio bayesiano), tanto para picos agudos como anchos con una velocidad computacional más rápida, [23] consulte la comparación de referencia de las herramientas de llamada de picos de ChIP-seq de Thomas et al. (2017). [24]
Otro problema computacional relevante es el llamado de picos diferenciales, que identifica diferencias significativas en dos señales ChIP-seq de distintas condiciones biológicas. Los llamadores de picos diferenciales segmentan dos señales ChIP-seq e identifican picos diferenciales utilizando modelos ocultos de Markov . Ejemplos de llamadores de picos diferenciales de dos etapas son ChIPDiff [25] y ODIN. [26]
Para reducir los sitios espurios de ChIP-seq, se pueden utilizar múltiples controles experimentales para detectar los sitios de unión de un experimento de IP. Bay2Ctrls adopta un modelo bayesiano para integrar el control de entrada de ADN para el IP, el IP simulado y su control de entrada de ADN correspondiente para predecir los sitios de unión del IP. [27] Este enfoque es particularmente eficaz para muestras complejas, como organismos modelo completos. Además, el análisis indica que, para muestras complejas, los controles de IP simulados superan sustancialmente a los controles de entrada de ADN, probablemente debido a los genomas activos de las muestras. [27]
Véase también
Métodos similares
- Secuenciación CUT&RUN , escisión controlada dirigida por anticuerpos mediante nucleasa microcócica en lugar de ChIP, lo que permite una mejor relación señal/ruido durante la secuenciación.
- Secuenciación CUT&Tag , escisión controlada dirigida por anticuerpos por la transposasa Tn5 en lugar de ChIP, lo que permite una mejor relación señal-ruido durante la secuenciación.
- Sono-Seq , idéntico a ChIP-Seq pero omitiendo el paso de inmunoprecipitación.
- HITS-CLIP [28] [29] (también llamado CLIP-Seq ), para encontrar interacciones con ARN en lugar de ADN.
- PAR-CLIP , otro método para identificar los sitios de unión de las proteínas de unión al ARN celular (RBP).
- RIP-Chip , mismo objetivo y primeros pasos, pero no utiliza métodos de reticulación y utiliza microarrays en lugar de secuenciación
- SELEX , un método para encontrar una secuencia de unión de consenso
- Competition-ChIP , para medir la dinámica de reemplazo relativa en el ADN.
- ChiRP-Seq para medir ADN y proteínas unidas al ARN.
- ChIP-exo utiliza un tratamiento con exonucleasa para lograr una resolución de hasta un solo par de bases
- ChIP-nexus es una versión mejorada de ChIP-exo para lograr una resolución de hasta un solo par de bases.
- DRIP-seq utiliza el anticuerpo S9.6 para precipitar híbridos DND:RNA de tres cadenas llamados bucles R.
- TCP-seq , un método principalmente similar para medir la dinámica de la traducción del ARNm.
- Calling Cards, utiliza una transposasa para marcar la secuencia donde se une un factor de transcripción. [30]
Referencias
- ^ Muhammad, Isiaka Ibrahim; Kong, Sze Ling; Akmar Abdullah, Siti Nor; Munusamy, Umaiyal (25 de diciembre de 2019). "RNA-seq y ChIP-seq como enfoques complementarios para la comprensión del mecanismo regulador de la transcripción en plantas". Revista Internacional de Ciencias Moleculares . 21 (1): 167. doi : 10.3390/ijms21010167 . ISSN 1422-0067. PMC 6981605 . PMID 31881735.
- ^ Johnson DS, Mortazavi A, Myers RM, Wold B (junio de 2007). "Mapeo de todo el genoma de interacciones proteína-ADN in vivo" (PDF) . Science . 316 (5830): 1497–502. Bibcode :2007Sci...316.1497J. doi :10.1126/science.1141319. PMID 17540862. S2CID 519841.
- ^ "Secuenciación de IP de cromatina de todo el genoma (ChIP-Seq)" (PDF) . Illumina, Inc. 26 de noviembre de 2007.
- ^ Song, Lingyun; Crawford, Gregory E. (febrero de 2010). "DNase-seq: una técnica de alta resolución para mapear elementos reguladores de genes activos en todo el genoma de células de mamíferos". Protocolos de Cold Spring Harbor . 2010 (2): pdb.prot5384. doi :10.1101/pdb.prot5384. ISSN 1559-6095. PMC 3627383. PMID 20150147 .
- ^ Giresi, Paul G.; Kim, Jonghwan; McDaniell, Ryan M.; Iyer, Vishwanath R.; Lieb, Jason D. (junio de 2007). "FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) aísla elementos reguladores activos de la cromatina humana". Genome Research . 17 (6): 877–885. doi :10.1101/gr.5533506. ISSN 1088-9051. PMC 1891346 . PMID 17179217.
- ^ Kumar, Vibhor; Muratani, Masafumi; Rayan, Nirmala Arul; Kraus, Petra; Lufkin, Thomas; Ng, Huck Hui; Prabhakar, Shyam (julio de 2013). "Procesamiento uniforme y óptimo de señales de datos mapeados de secuenciación profunda". Nature Biotechnology . 31 (7): 615–622. doi : 10.1038/nbt.2596 . ISSN 1087-0156. PMID 23770639. S2CID 32510475.
- ^ abcd «Guía ChIP: aplicaciones epigenéticas | Abcam» www.abcam.com . Consultado el 2 de marzo de 2020 .
- ^ Kim TH, Dekker J (abril de 2018). "Enlace cruzado de formaldehído". Protocolos de Cold Spring Harbor . 2018 (4): pdb.prot082594. doi :10.1101/pdb.prot082594. PMID 29610357.
- ^ Kim TH, Dekker J (abril de 2018). "Preparación de cromatina reticulada para ChIP". Protocolos de Cold Spring Harbor . 2018 (4): pdb.prot082602. doi :10.1101/pdb.prot082602. PMID 29610358.
- ^ Park, Peter J. (octubre de 2009). "ChIP-seq: ventajas y desafíos de una tecnología en proceso de maduración". Nature Reviews. Genetics . 10 (10): 669–680. doi :10.1038/nrg2641. ISSN 1471-0064. PMC 3191340 . PMID 19736561.
- ^ ab Chen, Yiwen; Negre, Nicolas; Li, Qunhua; Mieczkowska, Joanna O.; Slattery, Matthew; Liu, Tao; Zhang, Yong; Kim, Tae-Kyung; He, Housheng Hansen; Zieba, Jennifer; Ruan, Yijun (junio de 2012). "Evaluación sistemática de los factores que influyen en la fidelidad de ChIP-seq". Nature Methods . 9 (6): 609–614. doi :10.1038/nmeth.1985. ISSN 1548-7105. PMC 3477507 . PMID 22522655.
- ^ Jung, Youngsook L.; Luquette, Lovelace J.; Ho, Joshua WK; Ferrari, Francesco; Tolstorukov, Michael; Minoda, Aki; Issner, Robbyn; Epstein, Charles B.; Karpen, Gary H.; Kuroda, Mitzi I.; Park, Peter J. (mayo de 2014). "Impacto de la profundidad de secuenciación en experimentos ChIP-seq". Investigación de ácidos nucleicos . 42 (9): e74. doi :10.1093/nar/gku178. ISSN 1362-4962. PMC 4027199 . PMID 24598259.
- ^ Ho, Joshua WK; Bishop, Eric; Karchenko, Peter V.; Nègre, Nicolas; White, Kevin P.; Park, Peter J. (28 de febrero de 2011). "ChIP-chip versus ChIP-seq: lecciones para el diseño experimental y el análisis de datos". BMC Genomics . 12 : 134. doi : 10.1186/1471-2164-12-134 . ISSN 1471-2164. PMC 3053263 . PMID 21356108.
- ^ Jothi, et al. (2008). "Identificación de sitios de unión de proteína-ADN in vivo en todo el genoma a partir de datos de ChIP-seq". Nucleic Acids Res . 36 (16): 5221–5231. doi : 10.1093/nar/gkn488 . PMC 2532738 . PMID 18684996.
- ^ Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. (enero de 2005). "Mapas genómicos y análisis comparativo de modificaciones de histonas en humanos y ratones". Cell . 120 (2): 169–81. doi : 10.1016/j.cell.2005.01.001 . PMID 15680324. S2CID 7193829.
- ^ "HeLa S3 de ATCC | Biocompare.com". www.biocompare.com . Consultado el 21 de marzo de 2020 .
- ^ Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, et al. (agosto de 2007). "Perfiles de todo el genoma de la asociación del ADN de STAT1 mediante inmunoprecipitación de cromatina y secuenciación masiva en paralelo". Nature Methods . 4 (8): 651–7. doi :10.1038/nmeth1068. PMID 17558387. S2CID 28531263.
- ^ Schmid CD, Bucher P (noviembre de 2007). "Los datos de ChIP-Seq revelan la arquitectura de los nucleosomas de los promotores humanos". Cell . 131 (5): 831–2, respuesta del autor 832–3. doi : 10.1016/j.cell.2007.11.017 . PMID 18045524. S2CID 29234049.
- ^ Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. (septiembre de 2010). "Identificación de potenciadores cardíacos débilmente conservados mediante ChIP-Seq". Nature Genetics . 42 (9): 806–10. doi :10.1038/ng.650. PMC 3138496 . PMID 20729851.
- ^ Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, et al. (febrero de 2011). "Características de unión de factores de transcripción diversas reveladas por ChIP-seq de todo el genoma en C. elegans". Genome Research . 21 (2): 245–54. doi :10.1101/gr.114587.110. PMC 3032928 . PMID 21177963.
- ^ Kumar V, Muratani M, Rayan NA, Kraus P, Lufkin T, Ng HH, Prabhakar S (julio de 2013). "Procesamiento uniforme y óptimo de señales de datos mapeados de secuenciación profunda". Nature Biotechnology . 31 (7): 615–22. doi : 10.1038/nbt.2596 . PMID 23770639.
- ^ Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. (2008). "Análisis basado en modelos de ChIP-Seq (MACS)". Genome Biology . 9 (9): R137. doi : 10.1186/gb-2008-9-9-r137 . PMC 2592715 . PMID 18798982.
- ^ Xing H, Mo Y, Liao W, Zhang MQ (2012). "Localización de la unión proteína-ADN y modificación de histonas en todo el genoma mediante un método de punto de cambio bayesiano con datos de ChIP-seq". PLOS Comput Biol . 8 (7): e1002613. Bibcode :2012PLSCB...8E2613X. doi : 10.1371/journal.pcbi.1002613 . PMC 5429005 . PMID 22844240.
- ^ Thomas R, Thomas S, Holloway AK, Pollard KS (2017). "Características que definen los mejores algoritmos de llamada de picos de ChIP-seq". Brief Bioinform . 18 (3): 441–450. doi :10.1093/bib/bbw035. PMC 5429005 . PMID 27169896.
- ^ Xu H, Wei CL, Lin F, Sung WK (octubre de 2008). "Un enfoque HMM para la identificación de sitios de modificación diferencial de histonas en todo el genoma a partir de datos de ChIP-seq". Bioinformática . 24 (20): 2344–9. doi : 10.1093/bioinformatics/btn402 . PMID 18667444.
- ^ Allhoff M, Seré K, Chauvistré H, Lin Q, Zenke M, Costa IG (diciembre de 2014). "Detección de picos diferenciales en señales ChIP-seq con ODIN". Bioinformática . 30 (24): 3467–75. doi :10.1093/bioinformatics/btu722. PMID 25371479.
- ^ ab Xu, Jinrui; Kudron, Michelle M; Victorsen, Alec; Gao, Jiahao; Ammouri, Haneen N; Navarro, Fabio CP; Gevirtzman, Louis; Waterston, Robert H; White, Kevin P; Reinke, Valerie; Gerstein, Mark (21 de diciembre de 2020). "Simulacro o no: una comparación exhaustiva de IP simulada y entrada de ADN para ChIP-seq". Investigación de ácidos nucleicos . 49 (3): gkaa1155. doi : 10.1093/nar/gkaa1155 . ISSN 0305-1048. PMC 7897498 . PMID 33347581.
- ^ Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, et al. (noviembre de 2008). "HITS-CLIP ofrece información genómica sobre el procesamiento alternativo del ARN en el cerebro". Nature . 456 (7221): 464–9. Bibcode :2008Natur.456..464L. doi :10.1038/nature07488. PMC 2597294 . PMID 18978773.
- ^ Darnell RB (2010). "HITS-CLIP: vistas panorámicas de la regulación proteína-ARN en células vivas". Wiley Interdisciplinary Reviews. ARN . 1 (2): 266–286. doi :10.1002/wrna.31. PMC 3222227 . PMID 21935890.
- ^ Wang H, Mayhew D, Chen X, Johnston M, Mitra RD (mayo de 2011). "Las tarjetas de visita permiten la identificación multiplexada de los objetivos genómicos de las proteínas de unión al ADN". Genome Research . 21 (5): 748–55. doi :10.1101/gr.114850.110. PMC 3083092 . PMID 21471402.
Enlaces externos
- Catálogo ReMap: un análisis ChIP-Seq integrador y uniforme de elementos reguladores de más de 2800 conjuntos de datos ChIP-seq, que proporciona un catálogo de 80 millones de picos de 485 reguladores de la transcripción. [1]
- Base de datos ChIPBase: una base de datos para explorar mapas de unión de factores de transcripción a partir de datos ChIP-Seq . Proporciona el conjunto de datos ChIP-Seq más completo para diversos tipos de células y tejidos y condiciones.
- Base de datos y herramienta de análisis GeneProf: GeneProf es un entorno de análisis de libre acceso y fácil de usar para datos de ChIP-seq y RNA-seq y viene con una gran base de datos de experimentos públicos ya analizados, por ejemplo, para la unión de factores de transcripción y modificaciones de histonas.
- Llamada de pico diferencial Archivado el 15 de mayo de 2021 en Wayback Machine : Tutorial para llamadas de pico diferencial con ODIN.
- Análisis bioinformático de datos ChIP-seq: análisis exhaustivo de datos ChIP-seq. [2]
- KLTepigenome: descubrimiento de variabilidad correlacionada en conjuntos de datos epigenómicos utilizando la transformada Karhunen-Loeve.
- SignalSpider: una herramienta para el descubrimiento de patrones probabilísticos en múltiples perfiles de señales ChIP-Seq normalizados
- FullSignalRanker: una herramienta para la regresión y la predicción de picos en múltiples perfiles de señales ChIP-Seq normalizados
- ^ Chèneby J, Gheorghe M, Artufel M, Mathelier A, Ballester B (enero de 2018). "ReMap 2018: un atlas actualizado de regiones reguladoras a partir de un análisis integrador de experimentos de ChIP-seq de unión al ADN". Investigación de ácidos nucleicos . 46 (D1): D267–D275. doi :10.1093/nar/gkx1092. PMC 5753247 . PMID 29126285.
- ^ Bailey T, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, et al. (2013). "Pautas prácticas para el análisis exhaustivo de datos de ChIP-seq". PLOS Computational Biology . 9 (11): e1003326. Bibcode :2013PLSCB...9E3326B. doi : 10.1371/journal.pcbi.1003326 . PMC 3828144 . PMID 24244136.